Coronavirus 2019-nCoV: la più grande meta-analisi di tutti i genomi sequenziati
Realizzata all’Università di Bologna, conferma l’origine del virus nei pipistrelli e mostra una bassa eterogeneità: il virus è poco mutabile. Ma individua anche un punto di elevata variabilità
Lo studio ha analizzato i genomi dei 56 coronavirus 2019-nCoV finora sequenziati da vari laboratori nel mondo (Immagine: Scientific Animations)
La più grande meta-analisi realizzata di tutti i genomi finora sequenziati del coronavirus 2019-nCoV conferma la sua origine nei pipistrelli e mostra una bassa eterogeneità: il virus è poco mutabile. Al tempo stesso è stato però individuato un punto di elevata variabilità nelle proteine del virus, con l’esistenza di due sottotipi virali. Lo studio, pubblicato sul Journal of Medical Virology, è stato guidato da Federico M. Giorgi, ricercatore di bioinformatica al Dipartimento di Farmacia e Biotecnologie dell’Università di Bologna.
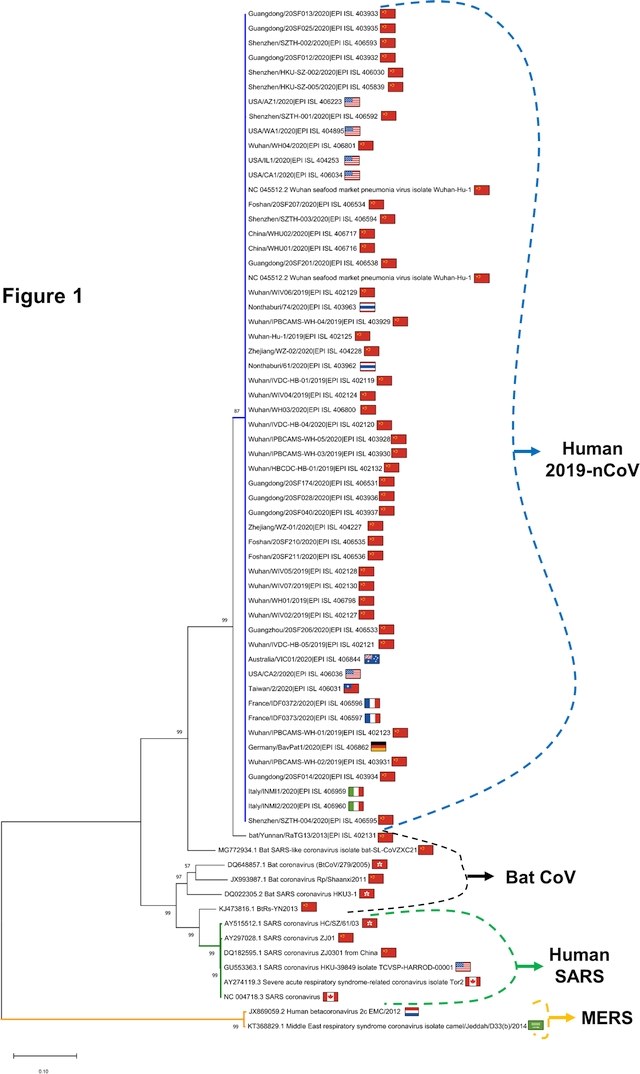
I dati dell'Organizzazione Mondiale della Sanità indicano che il coronavirus 2019-nCoV ha infettato fino ad oggi 24.554 persone, con 492 decessi confermati. Questo nuovo studio ha analizzato i genomi dei 56 coronavirus finora sequenziati da vari laboratori nel mondo, inclusi quelli derivanti dai due pazienti ricoverati in Italia, all'Istituto nazionale per le malattie infettive Lazzaro Spallanzani: si tratta dello studio più esteso di genomica comparativa per questo nuovo virus finora realizzato.
I ricercatori hanno confermato la probabile origine del coronavirus da una variante animale: il parente più stretto dei virus isolati in queste settimane corrisponde infatti alla sequenza EPI_ISL_402131 di un coronavirus di Rhinolophus affinis, un pipistrello asiatico di medie dimensioni, rinvenuto nella provincia dello Yunnan (Cina). Il genoma del nuovo coronavirus umano condivide almeno il 96,2% di identità con il suo probabile progenitore nel pipistrello, mentre si discosta molto di più dal genoma del virus umano responsabile della SARS (Severe Acute Respiratory Syndrome), con una somiglianza dell’80.3%.
I ricercatori hanno inoltre scoperto che tutti i coronavirus umani sequenziati fino ad oggi sono molto simili fra di loro, anche se provenienti da regioni diverse della Cina e del mondo: tutti i genomi ottenuti dai pazienti infettati dall’inizio dell’epidemia condividono un’identità di sequenza superiore al 99%. “Il virus è poco eterogeneo e mutabile: un dato ottimistico”, spiega Federico M. Giorgi. “Il fatto che la popolazione virale sia uniforme ci dice che un’eventuale terapia farmacologica dovrebbe funzionare su tutti”.
Lo studio ha però anche identificato per la prima volta un singolo punto di elevata variabilità nelle proteine del virus, con l’esistenza di due sottotipi virali. Questi differiscono per un singolo aminoacido in grado di cambiare sequenza e struttura nella proteina accessoria ORF8, una componente del virus che non è ancora stata caratterizzata.
Lo studio è stato pubblicato sul Journal of Medical Virology con il titolo “Genomic variance of the 2019-nCoV coronavirus”. Gli autori sono Federico M. Giorgi, ricercatore al Dipartimento di Farmacia e Biotecnologie dell’Università di Bologna, e Carmine Ceraolo, studente della laurea internazionale in Genomics dell’Università di Bologna.
Allegati